The overarching goal of our research is to develop accurate models of the complex processes underlying the function and regulation of cellular systems, particularly those whose malfunction contributes to disease. To accomplish this, we develop novel technologies for sophisticated analysis of diverse biological data, with a strong emphasis on the tight coupling of laboratory experiments, clinical data, and computational methods. With this approach, we uncover new knowledge about molecular pathways through integrative analysis and modeling of complex molecular-level changes captured via diverse functional genomics techniques, paving the way for a systems-level molecular view of complex disease. Our research interests fall roughly in the following broad categories:





Machine learning methods and applications
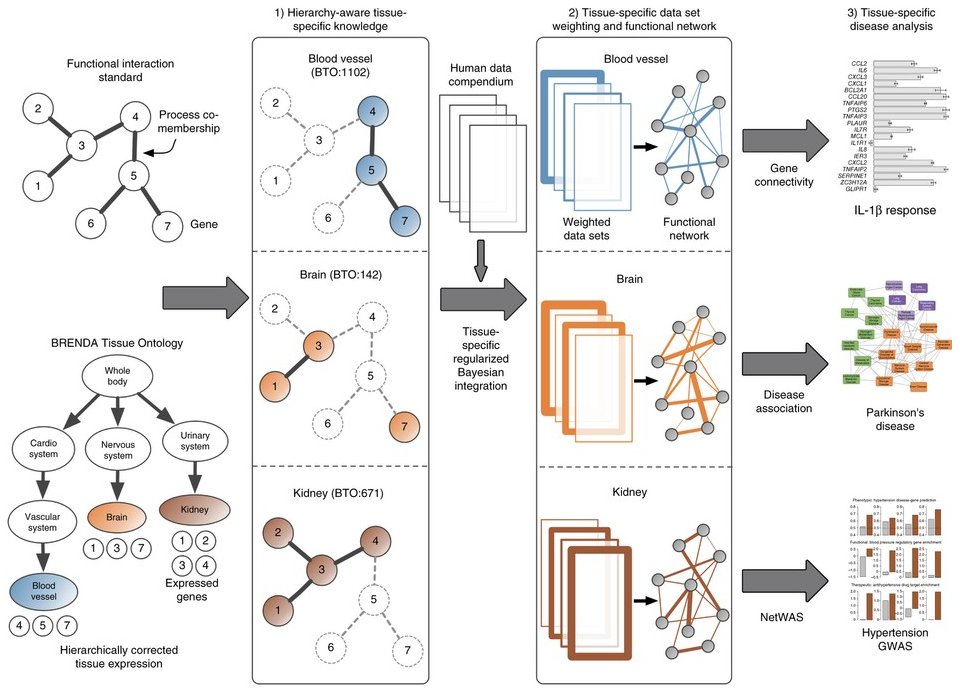
Regularized Bayesian integration based on tissue ontology. Our integration pipeline constructs tissue-specific functional interaction networks by (1) using tissue-specific knowledge to (2) identify and weight data sets by their tissue-relevant signal. (3) We demonstrate the capabilities of the networks by experimentally validating the gene connectivity scores (top), demonstrating that they identify disease associations (middle) and reprioritizing GWAS results (bottom).
The explosion of whole genome testing methodologies and the increasing push to make biological datasets publicly available has created a vast, unwieldy, repository of raw biological knowledge. Our work in this area pursues methods that combine these various data in a manner that reflects the data's reliability and biological accuracy.
Large-scale integrations of various data sources are often an effective first approximation of the functional landscape of a cell, and we have used these integrations in a wide range of applications, including predicting protein function, studying functional evolution, reprioritizing functional associations based on the results of genome-wide association studies, and establishing links to disease phenotypes.
We are also actively developing machine learning methods to better understand the function of noncoding regions in the genome. For example, our deep learning approach directly learns a regulatory sequence code from large-scale chromatin-profiling data, enabling prediction of chromatin effects of sequence alterations with single-nucleotide sensitivity.
Human disease and precision medicine
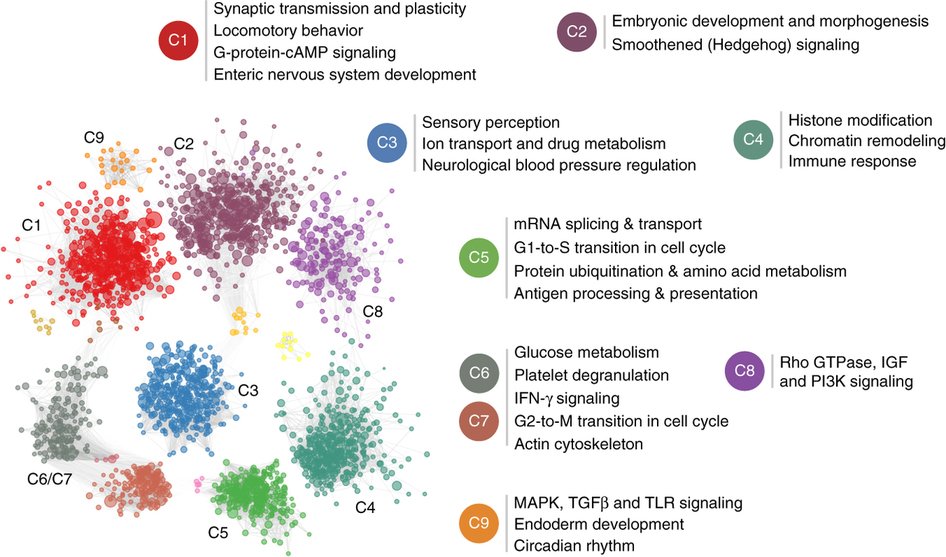
Autism-associated brain-specific functional modules. The network of brain-specific functional interactions among the top 2,500 ASD-associated genes were clustered using a shared-nearest-neighbor–based community-finding algorithm to elucidate several modules of genes (left). Nine of the clusters that contained 10 or more genes, labeled C1 through C9, were tested for functional enrichment using genes annotated to Gene Ontology biological process terms. Representative processes and pathways enriched within each cluster are presented here alongside the cluster label. Since C6 and C7 shared a number of strong links across the clusters, they were merged before calculating functional enrichment. The enriched functions provide a landscape of cellular functions potentially dysregulated by ASD-associated mutations.
An immense molecular complexity forms the foundation of human disease. Slight variations in the genetic makeup of individuals, as well as lifestyle and environmental factors, drive a host of different processes that contribute to dysfunction. A better understanding of this underlying heterogeneity will allow us to develop therapies that can effectively treat individuals while minimizing undesirable side-effects that accompany the traditional one-size-fits-all treatment paradigm.
Leveraging the synergies between high-throughput genomics and quantitative genetics studies, as well as traditional experimental and clinical data, we develop systems-level models to unravel patient complexity and uncover the key changes at the cellular level that give rise to disease phenotypes. Within the context of disease, we are also looking to improve understanding of differing drug responses and decrease the likelihood of off-target effects. Working closely with collaborators, we analyze the mechanisms of a wide variety of disease manifestations, including hypertension, cancer, and an array of neurological disorders (e.g., autism, Parkinson’s, Alzheimer’s).Interactive public systems and data visualization
Effective visualization-based analysis is critical to unlocking the full potential of genomic data and supporting collaborative research that is commonplace in genomics. We strongly believe in the importance of making the methods we develop and results we generate accessible to the wider community. To this end, we build user-friendly interactive public systems for our methods whenever possible and continue to develop methodologies for improved data visualization.
Modeling multicellular complexity
Tissue- and cell-lineage-specific gene expression underlie the development, function, and maintenance of diverse cell types within an organism. Many diseases also occur in specific target tissues; thus, understanding tissue-specific regulation is key to understanding pathogenesis. A major focal point of our research is accounting for cell-lineage specificity in integrated models of gene expression, protein interactions, and regulation, even though high-throughput data are rarely resolved for specific tissue types or cell-lineages.
More specifically, cell-lineage specificity is closely linked with dynamic processes of cell type differentiation, cellular regeneration, development, and aging. Modeling these dynamic processes using high-throughput data is extremely difficult, with the dynamic facets of the problem adding complexity beyond the standard data size, noise, and heterogeneity challenges. We are developing methods to address these challenges, moving towards the ultimate goal of creating high-resolution predictive dynamic models of cellular systems.
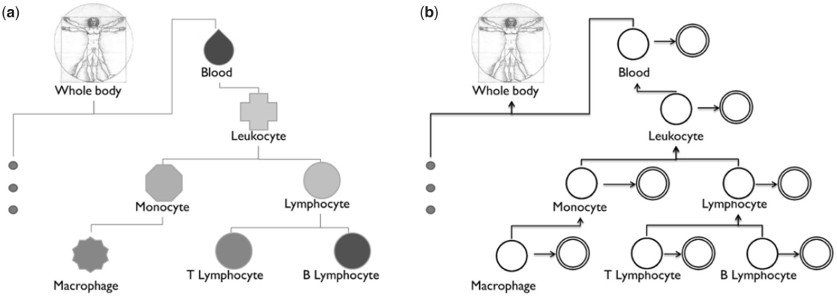
Leveraging the complex relationship between tissues and cell-types. (a) A small sub-tree of the BTO. This complexity has not yet been incorporated in tissue and cell-type-specific studies. (b) Our aggregation method uses this ontological structure to model the potential dependencies between individual cell-type models.
Example related publications:
- Understanding multicellular function and disease with human tissue-specific networks
- Ontology-aware classification of tissue and cell-type signals in gene expression profiles across platforms and technologies
- Global prediction of tissue-specific gene expression and context-dependent gene networks in Caenorhabditis elegans
Transcription regulation and epigenetics
Modern genome-scale experimental techniques can monitor diverse molecular events along multiple axes of cellular activity, such as transcriptional and post-transcriptional regulation, genetic and epigenetic changes, as well as various binding events. However, these high-throughput measurements are noisy and generally not comprehensive with respect to either the molecular players (e.g., incomplete coverage of proteins or metabolites) or experimental conditions (e.g., after perturbations). Furthermore, existing knowledge of detailed pathway mechanisms is incomplete, which severely limits the applicability of existing computational methods.
To develop a better understanding of the complexities of gene regulation amidst these challenges, we have developed machine learning methods to untangle the functional and spatial organization of chromatin code, as well as parse the regulatory code in noncoding sequences. Ultimately, we hope to provide a detailed, context-specific map of genetic regulatory pathways through integrative modeling of diverse genetic (e.g., sequence, expression) and epigenetic (e.g., nucleosomes, histone markers) signals.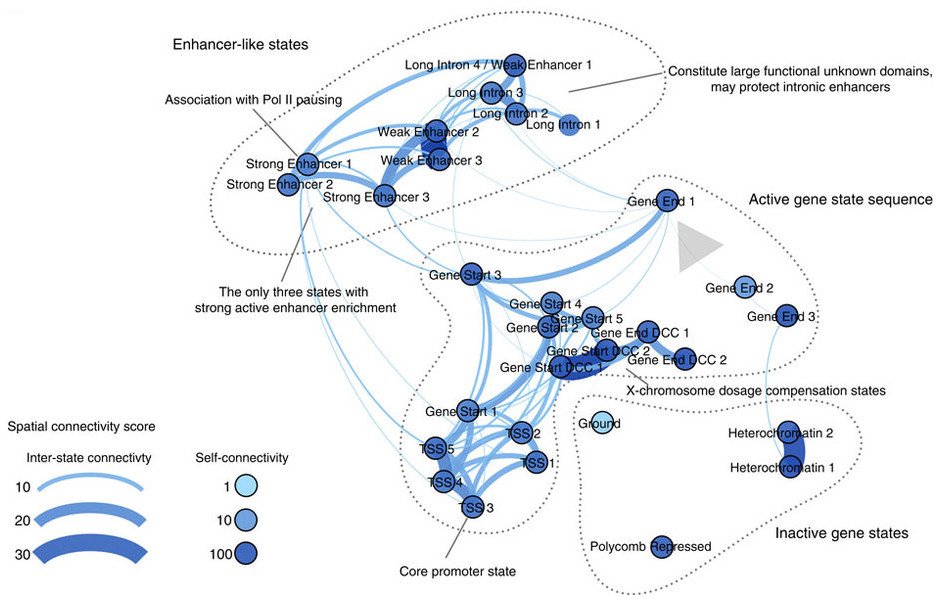
Discovery of chromatin states from the chromatin code energy landscape. Overview of the spatial and functional organization of the chromatin states discovered by our approach. The major groups of chromatin states include the following: (1) the enhancer-like states that include distinct functional groups of states carrying the widely used active enhancer marks H3K27ac and H3K4me1, (2) the active gene state sequence, for which we indicate the 5′→3′ sequence of states with a grey arrow, and (3) the inactive gene states. Significant findings about specific chromatin states are remarked. Edge width and colour indicates spatial connectivity score between chromatin states; a score of 30 indicates that the frequency of observing the two states in question being adjacent to each other in the genome is 30 times higher than expected by chance. Node colour darkness indicates the level of spatial connectivity score of the state to itself; most states appear very stable as indicated by the high self-connectivity score.
Example related publications:
- Predicting the effects of noncoding variants with deep learning-based sequence model
- Probabilistic modelling of chromatin code landscape reveals functional diversity of enhancer-like chromatin states
- Tissue-aware data integration approach for the inference of pathway interactions in metazoan organisms
Cross-species translational research
The interrogation of gene function in model organisms has vastly improved the understanding of human disease, in large part because core biological processes are evolutionarily conserved. However, while sequence similarity has been useful for transposition of information about gene function from one organism to another, inference of the role of a gene in the context of broader biological processes based on sequence similarity alone is generally less successful and fails to leverage the massive knowledge base that has been accumulated. Thus, we have developed robust network-based computational methods for identifying functionally analogous orthologs based on a combination of genome-scale datasets and sequence similarity.
Beyond this challenge, there remains the open problem of identifying endophenotypes across species. As with most complex diseases and phenotypes, the mapping of biological functions across species is not obvious even for experts, as it requires understanding of the processes underlying the disease and phenotype at a molecular, cellular, and organ level in each organism. For example, zebrafish is a common model for cardiovascular processes, but which pressure-related phenotype is most appropriate to study hypertension?
The untapped wealth of molecular-level information in genomic data available across multiple species provides us with the tools to address this challenge. This requires sophisticated data integration and analysis methods that can extract functional signals to map genes, processes, phenotypes and diseases across organisms in an unbiased, large-scale way. We have already made substantial progress on developing such methods, which utilize curated information to train our machine learning approaches to make novel discoveries. Ultimately, we believe that combining an organismal approach with molecular and cellular data shared across species will enable comparative ‘phenomics’ on a scale comparable with genomics.
Example related publications:
- An integrative tissue-network approach to identify and test human disease genes
- Functional Knowledge Transfer for High-accuracy Prediction of Under-studied Biological Processes
- Accurate Quantification of Functional Analogy among Close Homologs
- FNTM: a server for predicting functional networks of tissues in mouse